UMUM
Prosedur disolusi memerlukan suatu alat, media disolusi dan kondisi uji yang memberikan metode diskriminatif (mampu membedakan), sesuai ketegaran dan keberulangannya untuk dilakukan penetapan dari hari ke hari dan mampu dilakukan antar laboratorium.
Kriteria keberterimaan harus dapat mewakili beberapa bets dengan komposisi nominal yang sama dan proses pembuatannya, termasuk bets yang digunakan dalam penelitian dan mewakili kinerja dalam studi stabilitas.
Prosedur harus mampu membedakan dengan tepat perubahan bermakna dalam komposisi atau proses pembuatan yang mungkin dapat mempengaruhi kinerja in-vivo. Mungkin juga prosedur menunjukkan perbedaan antar bets ketika tidak ada perbedaan bermakna yang diamati dalam in vivo. Situasi ini memerlukan evaluasi saksama apakah prosedur terlalu sensitif atau memang mampu membedakan dengan tepat. Menilai hasil dari beberapa bets yang memperlihatkan variabilitas khas dalam parameter komposisi dan pembuatan, dapat membantu dalam evaluasi ini. Kadang-kadang merupakan hal yang berharga, untuk secara sengaja membuat variasi parameter pembuatan, seperti lubrikasi, waktu pencampuran, gaya tekan, atau parameter pengeringan, untuk lebih lanjut mencirikan kemampuan pembedaan dari prosedur.
Dengan mempertimbangkan stabilitas, uji disolusi harus menunjukkan perubahan yang sesuai terhadap sediaan obat dari waktu ke waktu yang disebabkan oleh suhu, kelembaban, fotosensitivitas dan tekanan lainnya.
Suatu uji yang dirancang dengan baik, harus memberikan data yang tidak terlalu bervariasi dan tidak dikaitkan dengan masalah stabilitas yang bermakna dari larutan analitik. Variabilitas yang tinggi dari hasil uji, menyebabkan sukar untuk melakukan identifikasi kecenderungan atau efek dari perubahan formulasi. Hasil disolusi dapat sangat bervariasi jika simpangan baku relatif (SBR) lebih besar dari 20% pada titik waktu tidak lebih dari 10 menit atau kurang dan (SBR) lebih besar 10% pada titik waktu selanjutnya. Namun, kebanyakan hasil disolusi menunjukkan variabilitas yang kurang dari nilai tersebut diatas. Sumber variabilitas harus diinvestigasi dan harus dilakukan usaha untuk mengurangi variabilitas, bila memungkinkan. Dua penyebab yang paling mungkin adalah formulasi (contoh: zat aktif, zat tambahan, atau proses pembuatan) dan peralatan yang berhubungan dengan prosedur uji (contoh: pengerucutan coning, tablet melekat pada dinding labu disolusi atau kawat keranjang). Pengamatan visual sering berguna untuk memahami sumber variabilitas dan apakah uji disolusi itu sendiri berkontribusi terhadap variabilitas. Hasil yang menyimpang dapat terjadi, setiap kali kandungan sediaan tidak bebas terdispersi di seluruh labu secara seragam. Tergantung pada masalahnya, biasanya dilakukan perbaikan termasuk mengganti tipe alat, kecepatan pengadukan atau awaudara; pertimbangan dan/atau pemeriksaan tipe singker; dan perubahan komposisi media. Modifikasi alat mungkin juga berguna dengan alasan yang tepat dan divalidasi.
Banyak penyebab variabilitas dapat ditemukan dalam formulasi dan proses pembuatan. Sebagai contoh, keseragaman sediaan yang buruk, ketidakkonsistenan proses, reaksi yang terjadi pada kecepatan yang berbeda selama disolusi, interaksi zat tambahan atau interferensi/gangguan, salut film, penuaan cangkang kapsul, dan pengerasan atau pelunakan bentuk sediaan pada stabilitas dapat menjadi sumber variabilitas dan interferensi. Selama pengujian rutin produk, variabilitas diluar rentang yang diharapkan harus diinvestigasi dari sudut analisis, formulasi dan proses pembuatan.
MEDIA
Data fisika dan kimia untuk bahan obat dan sediaan perlu ditentukan sebelum memilih media disolusi. Dua kunci sifat obat adalah kelarutan dan tingkat stabilitas larutan obat sebagai fungsi dari nilai pH. Ketika memilih komposisi media, pengaruh dapar, nilai pH, dan surfaktan pada kelarutan dan stabilitas obat perlu dievaluasi. Sifat utama dari sediaan yang mempengaruhi disolusi, termasuk mekanisme pelepasan (segera, tunda, atau modifikasi) dan kecepatan disintegrasi yang dipengaruhi oleh kekerasan, friabilitas/kerapuhan, adanya bahan peningkat kelarutan, dan adanya zat tambahan lainnya.
Pada umumnya, ketika mengembangkan prosedur disolusi, tujuan utama adalah memperoleh kondisi tenggelam (sink condition) yang didefinisikan sebagai volume media minimal tiga kali yang diperlukan untuk membentuk suatu larutan jenuh bahan obat. Pada saat kondisi tenggelam sudah terbentuk, hasil disolusi akan mencerminkan sifat bentuk sediaan. Suatu media yang gagal membentuk kondisi tenggelam, mungkin dapat diterima jika terbukti lebih mampu menunjukkan pembedaan atau memberikan alasan lain yang tepat.
Menggunakan campuran pelarut organik - pelarut berair sebagai media disolusi tidak dianjurkan; akan tetapi media jenis ini dapat diterima dengan alasan yang tepat.
Air murni sering digunakan sebagai media disolusi, tetapi tidak ideal karena beberapa alasan. Pertama, kualitas air dapat beragam tergantung pada sumber air, dan nilai pH air yang tidak dapat dikendalikan. Kedua, nilai pH dapat beragam dari hari ke hari dan dapat juga berubah selama pengujian, tergantung zat aktif dan zat tambahan. Terlepas dari keterbatasan ini, air tidak mahal, siap tersedia, mudah dibuang, secara ekologi dapat diterima, dan sesuai untuk produk dengan kecepatan pelepasan yang tidak tergantung pada nilai pH media.
Karakteristik disolusi dari formulasi oral harus dievaluasi dalam rentang pH fisiologis 1,2 sampai 6,8 (1,2 sampai 7,5 untuk formulasi sediaan lepas-modifikasi). Selama pengembangan metode, mengukur pH sebelum dan sesudah pengujian dapat berguna untuk mengetahui apakah pH berubah selama pengujian. Pemilihan kondisi metode yang paling tepat untuk pengujian rutin berdasarkan pada kapabilitas pembeda, ketegaran (ruggeness), stabilitas analit dalam media uji dan relevansi terhadap kinerja in vivo, jika memungkinkan.
Jenis media untuk disolusi dapat termasuk berikut (tidak terdaftar berdasarkan rekomendasi): asam hidroklorida encer, dapar dalam rentang pH fisiologis 1,2 sampai 7,5; cairan lambung buatan atau cairan usus buatan (dengan atau tanpa enzim) air, dan surfaktan (dengan atau tanpa asam atau dapar) seperti polisorbat 80, natrium lauril sulfat dan garam empedu.
Molaritas dapar dan asam yang digunakan dapat mempengaruhi efek melarutkan dan faktor ini mungkin dapat dievaluasi.
Untuk senyawa dengan kelarutan tinggi dan permeabilitas tinggi, pemilihan media dan alat dapat berpengaruh.
Untuk senyawa yang sangat buruk kelarutannya, larutan air mengandung persentase suatu surfaktan (contoh: natrium lauril sulfat, polisorbat, laurildimetilamin oksida) dapat digunakan untuk menambah kelarutan obat. Kebutuhan surfaktan dan konsentrasi yang digunakan dapat dibenarkan dengan menunjukkan profil pada beberapa konsentrasi yang berbeda. Surfaktan dapat digunakan sebagai bahan pembasah atau untuk melarutkan bahan obat.
Volume
Umumnya, untuk pengaduk keranjang dan dayung, volume media disolusi adalah 500 mL sampai 1000 mL. Volume yang paling umum adalah 900 mL. Volume dapat dinaikkan menjadi antara 2000 dan 4000 mL, menggunakan labu yang lebih besar dan tergantung pada konsentrasi dan kondisi tenggelam dari obat; diharapkan ada alasan untuk prosedur ini.
Awaudara
Peran awaudara pada media harus ditentukan, karena gelembung udara dapat mengganggu hasil uji, sebagai suatu penghalang untuk terjadi disolusi jika ada pada sediaan atau pada kawat keranjang. Selanjutnya, gelembung udara dapat menyebabkan partikel melekat pada alat dan dinding labu. Disamping itu, gelembung pada sediaan mungkin dapat meningkatkan daya apung, mengakibatkan meningkatnya laju disolusi atau menurunnya daerah permukaan yang tersedia sehingga menurunnya laju disolusi. Metode awaudara digambarkan pada catatan kaki pada Prosedur seperti tertera dalam Uji Disolusi <1231>. Langkah-langkah tersebut antara lain pemanasan media, penyaringan, dan menarik vakum untuk waktu singkat. Metode awaudara lainnya, tersedia dan secara rutin digunakan di industri. Media yang mengandung surfaktan biasanya tidak diawaudarakan karena hasil proses awaudara memberikan reaksi penyabunan yang berlebihan. Untuk menentukan apakah diperlukan awaudara terhadap media, hasil dari sampel disolusi dengan dan tanpa dilakukan awaudara terhadap media, harus dibandingkan.
Enzim
Penggunaan enzim pada media disolusi diperbolehkan sesuai yang tertera pada Uji Disolusi <1231> ketika disolusi gagal sebagai akibat dari cross-linking (silang antara) dengan kapsul gelatin dan produk salut gelatin.
Korelasi in vitro - in vivo
Media biorelevan adalah media yang memiliki beberapa korelasi terhadap kinerja sediaan secara in vivo. Pemilihan suatu media biorelevan berdasarkan pada (1) suatu pendekatan mekanik yang mempertimbangkan tempat absorpsi, jika diketahui, dan (2) apakah ’rate-limiting step’ dari absorpsi adalah disolusi atau permeabilitas senyawa. Dalam beberapa kasus, media biorelevan akan berbeda dari kondisi uji yang dipilih untuk pengujian yang ditetapkan dan juga titik waktu yang berbeda. Jika senyawa melarut cepat di lambung dan permeabilitas tinggi, maka waktu pengosongan lambung mungkin merupakan ’rate-limiting step’ dari absorpsi. Dalam hal ini, uji disolusi seharusnya menunjukkan bahwa obat dilepaskan dengan cepat pada kondisi lambung (asam). Disamping itu, jika disolusi terjadi terutama pada saluran pencernaan (contoh: untuk yang larut buruk, asam lemah), rentang pH lebih tinggi (contoh: cairan usus buatan dengan pH 6,8) mungkin lebih sesuai. Pada kondisi makan dan berpuasa mungkin juga memiliki efek bermakna terhadap absorpsi atau kelarutan senyawa. Komposisi media untuk kondisi makan dan berpuasa dapat ditemukan pada literatur. Media ini menggambarkan perubahan pH, konsentrasi empedu, dan osmolaritas setelah makan. Oleh karena itu terdapat perbedaan komposisi dari media kompendial. Media tersebut terutama digunakan untuk menetapkan korelasi in vitro - in vivo selama pengembangan formulasi dan untuk menilai efek potensial terhadap makanan dan tidak direncanakan untuk tujuan kontrol kualitas. Untuk tujuan kontrol kualitas, penggantian surfaktan dari alam (komponen empedu) dengan surfaktan buatan yang tepat, diperbolehkan dan dianjurkan karena mahalnya biaya bahan dari alam dan persiapan intensif dari media biorelevan.
ALAT/PENGADUK
Alat
Pemilihan alat berdasarkan pada pengetahuan rancangan formulasi dan praktek aspek kinerja bentuk sediaan pada sistem uji in vitro. Untuk bentuk sediaan padat oral, alat tipe 1 dan tipe 2 yang paling sering digunakan.
Ketika alat tipe 1 atau tipe 2 tidak sesuai, alat lain mungkin dapat digunakan. Alat tipe 3 (Reciprocating Cylinder) sudah digunakan untuk bentuk sediaan ‘bead-type modified-release’. Alat tipe 4 (Flow-Through Cell) dapat memberikan keuntungan untuk bentuk sediaan lepas-modifikasi yang mengandung bahan aktif dengan kelarutan terbatas. Disamping itu, alat tipe 3 atau tipe 4 memiliki kegunaan untuk kapsul gelatin lunak, ’bead product’, supositoria atau obat dengan kelarutan rendah. Alat tipe 5 ( Paddle over Disk) dan alat tipe 6 (Rotating Cylinder) digunakan untuk evaluasi dan uji dari bentuk sediaan transdermal. Alat tipe 7 (Reciprocating Holder) digunakan untuk bentuk sediaan oral non disintegrasi lepas-modifikasi, seperti pada untuk bentuk sediaan transdermal.
Beberapa perubahan dapat dibuat pada alat; contoh: ukuran mesh suatu keranjang dengan ukuran selain 40 mesh (antara lain 10, 20, 80 mesh) dapat digunakan ketika kebutuhan tersebut jelas didokumentasikan oleh data pendukung. Jika teredia berbagai ukuran mesh yang dipersyaratkan FI, bahan keranjang dengan dimensi metrik terdekat harus digunakan. Perlu diperhatikan bahwa keranjang harus seragam dan memenuhi persyaratan dimensi, seperti tertera pada Uji Disolusi <1231>. Jika permukaan keranjang menjadi tersumbat selama disolusi formulasi kapsul atau tablet, dianjurkan untuk menukar dengan metode pengaduk dayung. Volume dapat ditingkatkan dari 900 sampai 1000 mL dengan menggunakan labu 2000 sampai 4000 mL untuk membantu membentuk kondisi tenggelam pada obat dengan kelarutan rendah.
Suatu alat nonkompendial memiliki beberapa kegunaan dengan pembenaran yang tepat, kualifikasi dan pendokumentasian yang lebih unggul di atas alat standar. Contoh, suatu alat volume kecil dengan pengaduk dayung dan keranjang berukuran kecil (mini), dapat dipertimbangkan untuk produk dengan kekuatan dosis rendah. Botol berputar atau tabung statik (tabung stasioner berjaket tertutup dengan jaket air dan dilengkapi dengan suatu pengaduk magnetik) juga memiliki kegunaan untuk mikrosfer dan implan, labu yang tinggi untuk menghilangkan pengerucutan ‘coning’, dan modifikasi alat tipe 4 untuk bentuk sediaan khusus termasuk serbuk dan ’stents’.
Singker
Ketika singker digunakan, deskripsi singker harus diuraikan pada prosedur tertulis. Hal ini berguna untuk mengevaluasi perbedaan singker, mengenali bahwa singker dapat berpengaruh secara bermakna terhadap profil disolusi suatu sediaan. Ketika prosedur diganti, singker harus diduplikasi/ditiru sedekat mungkin dalam fasilitas berikutnya. Terdapat beberapa tipe singker yang tersedia di perdagangan. Metode untuk membuat sendiri singker menggunakan tangan, singker yang mirip dengan “beberapa putaran berbentuk spiral dari kawat” seperti yang diuraikan pada Alat tipe 2 (Alat Dayung) dalam Uji Disolusi <1231> digambarkan berikut ini.
Bahan - Gunakan kawat baja tahan karat 316 atau bahan iner lain, biasanya 0,032 inchi/20 gauge; dan silinder dengan diameter yang sesuai (antara lain gabus). Ukuran dapat dilihat pada tabel di bawah ini.
Tipe cangkang kapsul | Panjang kabel | Ukuran diameter (cm) | Nomor gabus |
#0, memanjang | 12 | 0,8 | 4 |
#1 dan #2 | 10 | 0,7 | 3 |
#3 dan #4 | 8 | 0,55 | 2 |
Prosedur Potong kawat dengan panjang tertentu, gulung mengelilingi silinder dengan ukuran yang sesuai, dan gunakan tang kecil untuk memelintirkan pada ujung kawat. Perhatikan ketika digunakan, karena ujung kawat mungkin kasar dan perlu dihaluskan.
Jika singker dibuat sendiri (menggunakan tangan), bahan singker dan penyusunan petunjuk prosedur harus didokumentasikan; jika menggunakan singker yang dibeli, nomor tipe harus dicatat.
Pengadukan
Untuk formulasi kapsul atau tablet lepas segera, alat tipe 1 (keranjang) pada 100 rpm atau alat tipe 2 (dayung) pada 50 atau 75 rpm paling banyak digunakan. Kecepatan pengadukan dan alat lain dapat diterima dengan pembenaran yang tepat.
Kecepatan di luar rentang 25 sampai 150 rpm biasanya tidak tepat karena ketidakkonsistensian hidrodinamik di bawah 25 rpm dan karena turbulensi di atas 150 rpm. Kecepatan pengadukan antara 25 dan 50 rpm umumnya dapat diterima untuk suspensi. Untuk bentuk sediaan yang menunjukkan pengerucutan ‘coning’ (‘mounding’) dengan menggunakan pengaduk dayung pada 50 rpm, pengerucutan ‘coning’ dapat dikurangi dengan meningkatkan kecepatan dayung sampai 75 rpm, kemudian mengurangi bagian-bagian kecil yang terlepas dan memperbaiki data. Dengan pembenaran, 100 rpm mungkin dapat digunakan, terutama untuk sediaan lepas lambat. Penurunan atau peningkatan kecepatan perputaran alat dapat dibenarkan jika profil yang lebih baik dalam menggambarkan kinerja in vivo dan/atau hasil metode menghasilkan pembedaan yang lebih baik tanpa mempengaruhi keberulangan metode.
Pemilihan pengadukan dan parameter lain untuk bentuk sediaan lepas-modifikasi mirip dengan sediaan lepas segera. Parameter tersebut harus sesuai dengan persyaratan dan spesifikasi yang tertera pada Uji Disolusi <1231> ketika alat telah dikalibrasi dengan tepat.
RANCANGAN PENGUJIAN
Titik Waktu
Untuk bentuk sediaan lepas segera, lamanya prosedur biasanya 30 sampai 60 menit. Umumnya spesifikasi satu titik waktu, cukup untuk persyaratan Farmakope. Konsep pada industri dan regulasi untuk kinerja dan kesesuaian produk mungkin memerlukan titik waktu tambahan yang juga mungkin diperlukan untuk pendaftaran atau persetujuan produk. Sejumlah titik waktu yang memadai, harus dipilih untuk cukup membedakan fase naik dan fase plato dari kurva disolusi. Menurut sistem klasifikasi biofarmasi (Biopharmaceutics Classification System), obat sangat larut dan sangat permiabel diformulasi untuk produk yang melarut dengan cepat, tidak perlu menggunakan perbandingan profil jika produk obat dapat menunjukkan pelepasan zat aktif obat tidak kurang dari 85% selama 15 menit. Untuk jenis produk ini, uji satu titik sudah cukup memadai. Bagaimanapun juga, kebanyakan produk tidak termasuk dalam kategori ini. Profil disolusi untuk produk lepas segera, biasanya menunjukkan suatu peningkatan bertahap mencapai 85% sampai 100% pada lebih kurang 30 sampai 45 menit. Kemudian titik waktu disolusi dalam rentang 15, 20, 30, 45 dan 60 menit biasanya untuk kebanyakan produk lepas segera. Untuk produk yang melarut dengan cepat, termasuk suspensi, informasi yang berguna mungkin diperoleh dari titik-titik waktu sebelumnya, misalnya 5 sampai 10 menit. Untuk produk yang melarut lebih lambat, titik waktu lebih dari 60 menit dapat digunakan. Titik-titik waktu uji disolusi untuk uji pada kompendia, biasanya ditetapkan berdasarkan suatu evaluasi data profil disolusi.
Titik waktu tak terbatas dapat berguna selama pengembangan penelitian. Untuk memperoleh waktu tak terbatas, kecepatan pengaduk dayung atau keranjang ditingkatkan pada akhir uji untuk periode berikutnya (biasanya 15 sampai 60 menit), setelah dilakukan uji dengan tambahan titik waktu. Meskipun tidak ada persyaratan untuk disolusi 100% pada profil disolusi, waktu tak terbatas dapat memberikan data yang dapat menambah keseragaman data dan memberikan informasi berguna tentang karakteristik formulasi selama awal pengembangan atau tentang ketidaktepatan metode.
Untuk bentuk sediaan lepas lambat, minimal tiga titik waktu uji yang dipilih untuk mengkarakterisasi profil pelepasan obat secara in vitro untuk memenuhi persyaratan farmakope. Penambahan titik waktu mungkin diperlukan untuk tujuan perizinan obat. Pada titik waktu awal, biasanya 1 sampai 2 jam, dipilih untuk menunjukkan adanya kemungkinan kecil dosis terbuang. Pada titik waktu menengah, dipilih untuk menunjukkan profil pelepasan bentuk sediaan secara in vitro, dan pada titik waktu akhir, pada dasarnya menunjukkan pelepasan obat secara keseluruhan. Titik-titik waktu uji dan spesifikasi biasanya ditetapkan berdasarkan pada evaluasi data profil pelepasan obat. Untuk produk yang mengandung lebih dari satu bahan aktif, pelepasan obat ditentukan untuk masing-masing bahan aktif.
Pengamatan
Pengamatan visual, pencatatan disolusi dan disintegrasi produk sangat berguna karena pola disolusi dan disintegrasi dapat menjadi indikasi dari variabel dalam formulasi atau proses produksi. Untuk melakukan pengamatan visual, sangat penting adanya pencahayaan yang tepat pada isi labu (dengan pertimbangan ada tidaknya fotodegradasi) dan visibilitas dari tangas. Mendokumentasikan pengamatan dengan menggambar sketsa dan mengambil foto atau video dapat menjadi pelajaran dan berguna untuk personil yang tidak dapat mengamati saat dilakukan uji disolusi. Pengamatan terutama berguna selama pengembangan metode dan optimasi formulasi. Berikut contoh pengamatan khas, namun tidak terbatas pada dibawah ini:
1. Tidak meratanya distribusi partikel pada labu. Hal ini dapat terjadi saat partikel melekat pada sisi labu, ketika ada pengerucutan ‘coning’ atau ‘mounding’ secara langsung pada alat, saat partikel mengambang pada permukaan media, saat tablet salut selaput menempel pada labu, dan/atau saat terbentuk ‘mounds’ di tengah-tengah.
2. Gelembung udara dalam labu atau pada alat atau pada sediaan. Kilau pada alat juga merupakan tanda adanya gelembung udara. Pengamatan ini, biasanya akan dilakukan ketika menilai kebutuhan untuk awaudara media.
3. Berputarnya sediaan atau sediaan terbentur pengaduk dayung.
4. Adhesi partikel terhadap pengaduk dayung atau bagian dalam dari keranjang, yang mungkin dapat diamati pada pergerakan pengadukan pada akhir proses disolusi.
5. ‘Pellicles’ atau formasi yang analog seperti kantong transparan atau karet, mengembangnya massa disekitar isi kapsul.
6. Adanya partikel besar atau potongan sediaan yang mengambang.
7. Pengamatan kecepatan disintegrasi (contoh, persentase penurunan unit dosis dalam jangka waktu tertentu)
8. Disintegrasi kompleks dari penyalutan yang dimodifikasi atau produk salut enterik. Contoh : terbuka sebagian dan membelah terpisah (seperti kulit kerang) atau terbukanya cangkang secara tidak sempurna disertai dengan pelepasan gelembung udara dan zat tambahan.
Sampling
Manual Sampling secara manual menggunakan siring dari plastik atau kaca, suatu kanula dari baja tahan karat yang biasanya melengkung untuk memungkinkan sampling pada labu, suatu penyaring dan/atau suatu pemegang penyaring. Posisi sampling harus disesuaikan dengan spesifikasi yang tertera pada Uji Disolusi <1231>
Autosampling Sampling secara otomatis (autosampling) adalah alternatif lain sampling secara manual yang berguna khususnya jika uji dilakukan pada beberapa titik waktu. Tetapi karena aturan pada laboratorium dalam menjalankan uji disolusi menggunakan sampling manual, maka autosampling memerlukan validasi terhadap sampling manual.
Ada banyak merk autosamplers, termasuk sistem semiotomatis dan otomatis penuh. Pemeriksaan kinerja yang dilakukan secara rutin, pembersihan dan perawatan seperti yang diuraikan pada prosedur operasional baku atau dokumen metrologi merupakan hal-hal yang berguna untuk pemakaian yang dapat dipercaya dari alat ini.
Beberapa alat dilengkapi dengan sampling melalui keranjang atau batang pengaduk dayung. Validasi yang tepat (antara lain menunjukkan kesetaraan terhadap hasil dengan prosedur sampling yang biasa dilakukan) mungkin diperlukan.
Gangguan hidrodinamika labu oleh alat pengambil sampel harus dipertimbangkan dan dilakukan validasi yang memadai untuk memastikan bahwa alat tidak menyebabkan perubahan yang bermakna terhadap laju disolusi.
Perbandingan prosedur manual dan otomatis harus dilakukan untuk mengevaluasi pergantian prosedur. Hal ini dapat dicapai dengan membandingkan data dari dua prosedur sampling secara terpisah atau dalam beberapa kasus, sampling dengan ke dua cara tersebut dilakukan pada labu yang sama. Hasil harus konsisten dengan persyaratan untuk presisi antara (diuraikan pada bagian Validasi) jika prosedur diganti.
Aspek lain dari validasi proses otomatisasi, dapat mencakup sisa-sisa residu obat, pengaruh alat (sampling secara simultan yang disebutkan diatas tidak sesuai dengan kasus ini), adsorpsi obat, dan siklus pembersihan dan/atau pembilasan.
Penyaring
Penyaringan sampel disolusi biasanya diperlukan untuk mencegah partikel obat yang tidak melarut, masuk ke dalam sampel analitik dan kemudian melarut. Penyaringan juga menghilangkan zat tambahan tidak larut yang mungkin menyebabkan latar belakang tinggi atau kekeruhan. Pembasahan awal dengan media terhadap penyaring mungkin diperlukan.
Penyaring dapat sejalan atau pada akhir alat pengambil sampel atau keduanya. Porositas penyaring dapat berkisar antara 0,45 sampai 70 µm. Jenis penyaring yang biasa digunakan adalah ‘depth’, cakram, dan ‘flow-through’. Jika gangguan zat tambahan tinggi, atau filtrat tampak berkabut, atau jika penyaring tersumbat, harus dievaluasi alternatif jenis penyaring atau porositas penyaring.
Adsorpsi obat pada penyaring perlu dievaluasi. Jika adsorpsi obat terjadi, jumlah filtrat awal yang dibuang, mungkin perlu ditambah. Jika hasil masih tidak sesuai, dapat dicari suatu bahan penyaring alternatif.
Validasi penyaring dapat dicapai dengan pembuatan larutan baku yang sesuai atau larutan sampel terdisolusi sempurna (antara lain, dibuat sebagai suatu sampel yang khas dalam suatu labu atau sampel dimasukkan ke dalam gelas piala dan diaduk dengan pengaduk magnetik selama 1 jam). Untuk larutan baku, bandingkan hasil larutan yang disaring (setelah membuang volume awal yang sesuai) terhadap larutan yang tidak disaring. Untuk larutan sampel, bandingkan hasil larutan yang disaring (setelah membuang volume awal yang tepat) terhadap larutan yang disentrifus tanpa disaring.
Sentrifugasi
Sentrifugasi sampel tidak dipilih karena disolusi dapat berlanjut dan karena mungkin ada gradien konsentrasi pada beningan. Pengecualian untuk senyawa yang mengadsorbsi pada semua penyaring yang umum digunakan.
Penetapan Kadar
Penetapan kadar yang biasa dilakukan untuk sampel disolusi adalah penetapan secara spektrofotometri atau Kromatografi Cair Kinerja Tinggi (KCKT). Metode analisis yang lebih disukai adalah penetapan secara spektrofotometri karena hasil dapat diperoleh lebih cepat, analisis lebih sederhana, dan pelarut yang digunakan lebih sedikit. Metode KCKT digunakan jika ada gangguan yang bermakna dari zat tambahan atau diantara obat pada formulasi untuk memperbaiki sensitivitas analitik dan/atau ketika analisis dapat diotomatisasi. Hal ini mungkin berguna dalam memperoleh data untuk obat dengan penetapan kadar yang mengindikasikan stabilitas (misalnya kromatogram KCKT) dalam media terpilih, bahkan jika penetapan kadar didasarkan pada metode spektrofotometri.
Validasi
Topik validasi yang diuraikan pada bagian ini adalah khas tetapi tidak semua inklusif. Elemen validasi dapat beragam, tergantung pada fase pengembangan atau kegunaan yang dimaksudkan untuk data. Kriteria keberterimaan dinyatakan hanya sebagai pedoman dan dapat berbeda untuk beberapa produk. Perusahaan seharusnya mendokumentasikan kriteria keberterimaan untuk produknya dalam prosedur operasional baku. Pertimbangan lain mungkin penting untuk bentuk sediaan khusus. Tingkat validasi tergantung pada fase pengembangan produk. Validasi lengkap dilaksanakan pada waktu dilakukan studi klinik fase III. Studi validasi harus menunjukkan variasi yang berhubungan dengan profil titik waktu yang berbeda. Untuk produk yang mengandung lebih dari satu bahan aktif obat, metode disolusi harus divalidasi untuk masing-masing bahan aktif.
Spesifisitas/Gangguan Plasebo
Sangat penting untuk menunjukkan bahwa hasil tidak semestinya dipengaruhi oleh komponen plasebo, zat aktif obat lain, atau degradasinya.
Plasebo terdiri dari semua zat tambahan dan penyalut (juga termasuk zat warna, singker, dan cangkang kapsul yang sesuai) tanpa bahan aktif. Gangguan plasebo dapat ditetapkan dengan menimbang sampel campuran plasebo dan melarutkan atau mendispersikan ke dalam media disolusi pada konsentrasi yang diketahui selama uji. Diharapkan uji ini dilakukan pada suhu 37°, bandingkan dengan baku 100% dengan rumus:
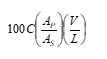
C adalah kadar baku dalam mg per mL; AP dan AS berturut-turut adalah serapan plasebo dan baku; V adalah volume media dalam mL; dan L adalah jumlah bahan aktif dalam mg yang tertera pada etiket. Gangguan tidak lebih dari 2%.
[Catatan Untuk produk lepas-lambat, plasebo dari bentuk sediaan akhir mungkin lebih tepat digunakan dari pada campuran, karena formulasi plasebo ini akan melepaskan beragam zat tambahan yang lebih menunjukan produk daripada campuran sederhana dari zat tambahan. Pada kasus ini, akan tepat jika mengevaluasi gangguan potensial pada beberapa titik sampling dalam profil pelepasan.]
Jika gangguan plasebo lebih dari 2%, maka modifikasi metode seperti (1) pemilihan panjang gelombang lain, (2) pengurangan garis dasar menggunakan panjang gelombang yang lebih panjang, atau (3) menggunakan KCKT, mungkin diperlukan untuk mencegah gangguan. Jika ada zat aktif obat lain atau degradasinya yang bermakna, perlu ditunjukkan bahwa hal tersebut tidak mempengaruhi hasil secara bermakna. Prosedur untuk melakukan hal ini adalah mengukur matrik dengan atau tanpa zat aktif obat lain atau degradasinya: gangguan tidak lebih dari 2%.
Linearitas dan Rentang
Linearitas dan rentang ditetapkan dengan membuat larutan obat, pada rentang kadar di bawah kadar terendah yang diharapkan hingga di atas kadar tertinggi selama pelepasan. Hal ini dapat dilakukan bersamaan dengan penetapan akurasi/perolehan kembali. Skema dapat diubah jika berbeda ukuran ‘flow-cell’ atau volume penyuntikan yang digunakan.
Jika memungkinkan, larutan dibuat dari satu larutan persediaan yang umum. Untuk kadar tertinggi, penetapan tidak boleh melebihi batas linieritas alat.
Pelarut organik dapat digunakan untuk meningkatkan kelarutan obat pada pembuatan larutan baku; akan tetapi tidak lebih dari 5% (v/v) pelarut organik dalam larutan akhir yang seharusnya digunakan, kecuali dilakukan validasi.
Linearitas dapat dihitung menggunakan program ‘least-squares regression’ yang sesuai. Koefisien korelasi kuadrat (r2 ? 0,98) menunjukkan linearitas, kemiringan harus tidak berbeda secara bermakna dari nol.
Akurasi / Perolehan kembali
Akurasi/perolehan kembali ditetapkan dengan membuat berbagai sampel yang mengandung obat dan kandungan lain yang ada dalam bentuk sediaan (antara lain zat tambahan, bahan penyalut, cangkang kapsul) pada rentang kadar di bawah kadar terendah yang diharapkan hingga di atas kadar tertinggi selama pelepasan.
Pada obat dengan kelarutan rendah, akurasi/perolehan kembali sebaiknya dilakukan dengan membuat larutan persediaan, dengan melarutkan bahan obat dalam sedikit pelarut organik (tidak lebih dari 5%) dan mengencerkan dengan media disolusi sampai kadar akhir. Sejumlah larutan persediaan yang setara dengan jumlah zat aktif yang tertera pada etiket dapat ditambahkan ke dalam labu menggantikan serbuk obat. Dengan cara yang sama, untuk obat dengan kekuatan sangat rendah, lebih baik membuat larutan persediaan daripada menimbang sejumlah sangat kecil bahan obat. Umumnya, perolehan kembali antara 95 dan 105% dari jumlah yang ditambahkan. Melakukan penggolongan matriks berbagai kadar dapat berguna pada tahap ini.
Kasus khusus untuk validasi adalah prosedur Tahap Asam yang diuraikan pada Sediaan Lepas Tunda dalam Uji Disolusi <1231>. Batas tidak lebih dari 10% harus divalidasi. Jika senyawa terurai dalam asam, percobaan validasi harus berdasarkan pada kenyataan ini.
Presisi
Keberulangan Keberulangan ditetapkan melalui pengukuran berulang larutan baku dan/atau larutan uji. Keberulangan dapat diukur dengan perhitungan (SBR) dari beberapa kali penyuntikan atau pembacaan spektrofotometri untuk masing-masing laruan baku atau dari akurasi atau linearitas data.
Presisi Antara (Intermediate precision) Presisi antara dapat dievaluasi untuk menetapkan pengaruh presisi prosedur analitik secara acak. Evaluasi ini dilakukan setelah pengembangan produk obat. Dengan presisi dapat menjelaskan rentang kekuatan produk. Variasi yang diteliti adalah hari, analis dan peralatan. Penggunaan rancangan matrik percobaan mendukung evaluasi presisi antara. Jika memungkinkan, presisi antara dapat dievaluasi menggunakan lot produk obat yang memiliki karakteristik baik dan keseragaman kandungan yang baik. Bila produk berkarakteristik baik tersebut tidak tersedia, plasebo dan bahan aktif dapat digunakan untuk mengidentifikasi presisi antara.
Profil disolusi pada sampel yang sama dapat dilakukan, setidaknya pada dua analis yang berbeda, tiap analis membuat larutan baku dan media. Analis menggunakan tangas disolusi, spektrofotometer atau alat KCKT (termasuk kolom) dan autosampler yang berbeda dan analis melakukan uji pada hari yang berbeda. Prosedur ini mungkin tidak perlu dilakukan untuk tiap kekuatan; sebagai gantinya, kekuatan tinggi dan rendah dapat diterima.
Kriteria keberterimaan adalah perbedaan pada nilai rata-rata antara hasil disolusi pada dua kondisi menggunakan kekuatan yang sama, tidak lebih 10% absolut pada titik waktu kurang dari 85% terlarut dan tidak lebih 5% untuk titik waktu diatas 85%. Kriteria keberterimaan untuk produk spesifik, serta batas dan uji statistik lain dapat digunakan.
Ketegaran (Robustness)
Evaluasi ketegaran untuk menilai efek yang dibuat kecil secara disengaja dengan mengubah kondisi disolusi, dilakukan selanjutnya pada saat pengembangan produk obat. Jumlah replikasi (3 atau 6) tergantung pada presisi antara.
Parameter dapat diubah, tergantung pada prosedur disolusi dan tipe analisis. Parameter tersebut termasuk komposisi media (antara lain kadar dapar atau surfaktan), pH, volume, kecepatan pengadukan dan suhu. Untuk analisis KCKT, parameter tersebut termasuk komposisi fase gerak (persentase larutan organik, kadar dapar, pH), laju alir, panjang gelombang, suhu kolom dan berbagai kolom dengan tipe yang sama. Untuk analisis secara spektrofotometri, panjang gelombang dapat diubah.
Stabilitas Larutan Baku dan Larutan Uji
Larutan baku disimpan pada kondisi yang terjamin stabilitasnya. Stabilitas baku dianalisis pada periode waktu yang ditentukan, menggunakan larutan baku yang dibuat baru, pada tiap interval waktu sebagai pembanding. Rentang yang dapat diterima untuk stabilitas larutan baku adalah antara 98% dan 102%.
Larutan uji disimpan pada suhu ruang, dianalisis pada periode waktu yang ditentukan, menggunakan respons larutan uji asli sebagai pembanding. Rentang yang dapat diterima untuk stabilitas larutan uji antara 98% dan 102% dibandingkan dengan analisis awal dari larutan uji. Jika larutan tidak stabil, hal yang dapat dipertimbangkan adalah suhu (lemari pendingin mungkin diperlukan), terlindung cahaya, dan wadah bahan (plastik atau kaca).
Pada prosedur perlu dinyatakan bahwa baku dan uji dianalisis dalam periode waktu dimana larutan tersebut stabil.
Analisis secara Spektrofotometri
Larutan uji dapat secara otomatis diukur pada spektrofotometer menggunakan pengisap otomatis (autosipper) dan ‘flow cell’. Pemeriksaan kinerja yang dilakukan secara rutin, pembersihan dan perawatan seperti yang diuraikan pada prosedur operasional baku atau dokumen metrologi merupakan hal-hal yang berguna untuk pemakaian yang dapat dipercaya dari alat ini. Biasanya digunakan sel dengan panjang pada rentang dari 0,02 sampai 1 cm. Posisi sel dan gelembung udara dapat menjadi sumber kesalahan. Sel dengan panjang lebih kecil digunakan untuk mencegah pengenceran larutan uji; bagaimanapun juga, linearitas yang dapat diterima dan kesalahan baku perlu ditunjukkan.
Selama analisis, biasanya larutan baku dibuat dan dianalisis hanya pada satu kadar, yaitu pada 100% (atau nilai Q yang dipilih) dari kekuatan dosis. Selama profil analisis, kadar lain dapat digunakan. Larutan blangko, baku, dan uji dapat dianalisis dalam serangkaian yang menghubungkan larutan uji dengan baku dan blangko, terutama pada awal dan akhir analisis.
Dalam kebayakan kasus, serapan rata-rata blangko media disolusi tidak lebih dari 1% terhadap baku. Nilai yang lebih tinggi dari 1% harus dievaluasi berdasarkan kasus per kasus. (SBR) untuk analisis ultra violet biasanya tidak lebih dari 2%.
Absorptivitas dihitung dengan membagi serapan rata-rata baku dengan kadar, dalam mg per mL, dibagi dengan panjang ‘flow cell’ dalam cm. Setelah semua data cukup terkumpul, dapat ditetapkan rentang absorptivitas yang dapat diterima untuk analit (menggunakan ‘flow cell’ yang sesuai). Nilai ini dapat digunakan untuk mengatasi penyimpangan data.
Serat optik sebagai sampling dan metode penetapan, dengan validasi yang tepat dapat menjadi sebuah pilihan.
Pemeriksaan spektrum ultra violet dari larutan obat mungkin berguna untuk memilih panjang gelombang optimum.
KCKT
Untuk analisis KCKT, dapat diperiksa kesesuaian antara media disolusi dan fase gerak, terutama jika diperlukan injektor volume besar (lebih dari 100 µl). Sampel biasanya dianalisis dengan KCKT menggunakan sebuah detektor spektrofotometrik dan penyuntik otomatis (auto-injector). Penyuntikan tunggal dari tiap labu, pada titik waktu dengan baku suatu sistem merupakan desain yang umum. Uji kesesuaian sistem mencakup sekurang-kurangnya waktu retensi dan ketepatan volume penyuntikan. Umumnya, pada analisis KCKT (SBR) tidak lebih dari 2% pada lima atau enam kali penyuntikan larutan baku. Tingkat baku biasanya pada 100% terhadap jumlah yang tertera pada etiket, terutama untuk analisis ’single-point’.
Pembuatan sampel plasebo untuk analisis KCKT dilakukan dengan cara yang sama seperti pada analisis secara spektrofotometri. Pemeriksaan kromatogram untuk puncak yang tereluasi pada waktu retensi yang sama seperti obat. Jika ada puncak lain, suntikkan larutan baku, dan bandingkan waktu retensinya. Jika waktu retensi terlalu dekat, tambahkan obat pada larutan plasebo. Kromatogram dapat juga diperoleh dari waktu tambahan menggunakan blangko (media disolusi), baku, dan larutan uji untuk mengidentifikasi bahan tereluasi terakhir yang mungkin mengganggu analisis berikutnya.
Dokumentasi validasi termasuk kromatogram atau spektra blangko media disolusi, larutan plasebo yang disaring, larutan baku, dan sampel disolusi yang disaring. Tidak adanya puncak pengganggu dalam kromatogram plasebo atau kurangnya serapan plasebo pada panjang gelombang analitik, menunjukkan spesivisitas.
Kriteria Keberterimaan
Kriteria keberterimaan untuk jumlah zat aktif terlarut, dinyatakan sebagai persentase jumlah yang tertera pada etiket (Q), dalam rentang 75% sampai 80% terlarut. Nilai Q yang lebih dari 80% tidak umum digunakan, karena perkiraan kebutuhan dibuat untuk rentang penetapan kadar dan keseragaman kandungan. Kriteria keberterimaan termasuk waktu uji yang biasa ditetapkan berdasarkan evaluasi data profil disolusi. Kriteria keberterimaan harus konsisten dengan data terdahulu, dan suatu harapan bahwa bets yang dapat diterima (antara lain tidak ada perbedaan yang bermakna dalam kinerja in vivo, komposisi, atau prosedur produksi) akan menghasilkan kesesuaian dengan kriteria keberterimaan.